Fourier- and Gábor Transform Analysis Methods for Highly Congested Mass Spectra of Heterogeneous Samples

Highly heterogeneous samples arise in many contexts, from biomolecular assemblies to co-polymers, and their electrospray ionization mass spectra can be extremely challenging to analyze by conventional means. Congestion in these mass spectra can be the result of many overlapping charge states with differing numbers of various repeated subunits or ligands, as well as chemical interference from salt clusters. The Prell Group develops analytical methods to facilitate analysis of highly congested spectra and applies them to study the physical properties of membrane protein-lipid assemblies, copolymers, and proteins and protein complexes electrosprayed from non-volatile salt buffers. A Python program for Fourier analysis of mass spectra with repeated subunits (lipids, monomers in polymers, etc.), which we call “iFAMS” (interactive Fourier-Transform Analysis for Mass Spectra), is now available at https://github.com/seanpatcleary/ifams. Please email Andy Swansiger (aswansig–AT–uoregon–DOT–edu) or see our recent article for further details. (If you use iFAMS, we kindly request that you cite this article in any publications or presentations resulting from it.) Upcoming features of iFAMS 6.3 include: automatic/batch process calibration for protein quantitation, Hough-Transform-based peak series detection, and applications to mass spectrometry-based tissue imaging.
Contact persons: Andy Swansiger, Kayd Meldrum, and Lily Miller
Funding: NSF CAREER CHE-1752994, Agilent University Relations Award.
Relating Protein Complex Unfolding and Dissociation to Native Structures: IonSPA
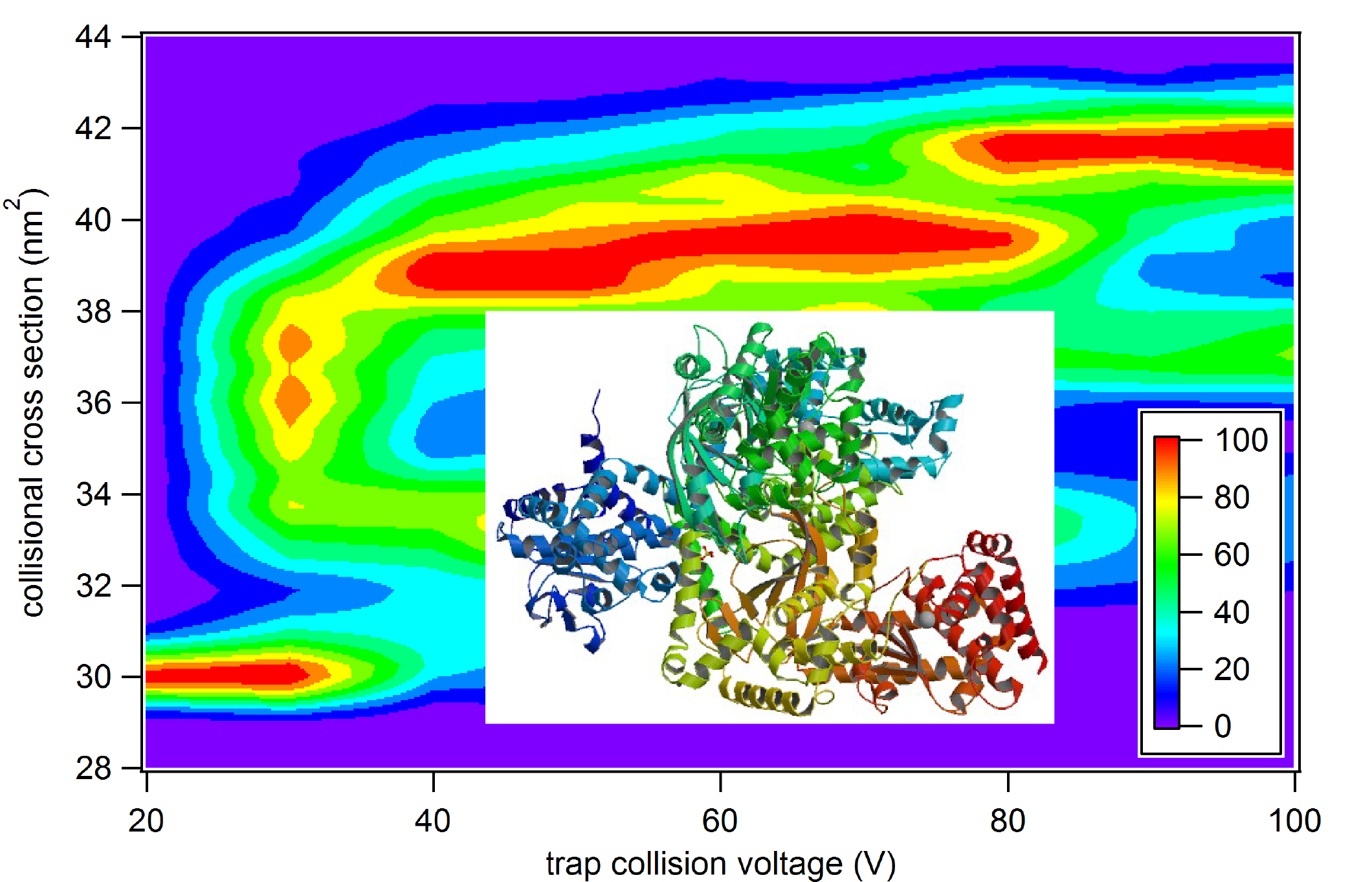 Native ion mobility-mass spectrometry, in combination with exciting new tools such as Surface Induced Dissociation, can be a powerful technique for revealing the quaternary and even tertiary structure of native-liked protein and protein complexes ionized gently from solution. The use of these tools to obtain quantitative information about interface geometry and structure is in its infancy, and we develop experimental, theoretical, and computational methodology to maximize the qualitative and quantitative information that can be learned with these methods. (Read our recent article here on quantitating energy deposition in CID and SID.) We are currently developing a software suite (Ion Simulations of the Physics of Activation: IonSPA) to enable researchers around the world to predict ion heating and cooling in various regions of common mass spectrometry instrumentation and, conversely, to determine ion thermochemistry from gas-phase measurements.
Native ion mobility-mass spectrometry, in combination with exciting new tools such as Surface Induced Dissociation, can be a powerful technique for revealing the quaternary and even tertiary structure of native-liked protein and protein complexes ionized gently from solution. The use of these tools to obtain quantitative information about interface geometry and structure is in its infancy, and we develop experimental, theoretical, and computational methodology to maximize the qualitative and quantitative information that can be learned with these methods. (Read our recent article here on quantitating energy deposition in CID and SID.) We are currently developing a software suite (Ion Simulations of the Physics of Activation: IonSPA) to enable researchers around the world to predict ion heating and cooling in various regions of common mass spectrometry instrumentation and, conversely, to determine ion thermochemistry from gas-phase measurements.
Translocation of endogenous and exogenous proteins across the membranes often requires the protein being translocated to undergo significant unfolding. We use gas-phase unfolding and dissociation (both collision-induced and surface-induced) combined with molecular dynamics simulations and state-of-the-art collisional cross section calculations to gain insights into the dynamics and energetics of unfolding, translocation, and dissociation processes.
Contact persons: Sam Shepherd, Dr. Micah Donor (now at George Fox University)
Funding: NIH R01GM141733 (PIs: Daniel Zuckerman, OHSU, and Elisar Barbar, Oregon State), Agilent University Relations Award, (previously) NIH R21AI125804
Quantitative Studies of Protein Unfolding and Dissociation
Native ion mobility-mass spectrometry (IM-MS), when coupled with gas-phase activation methods such as collision-induced unfolding (CIU) and surface-induced dissociation (SID), can probe the quaternary and tertiary structure of proteins and protein complexes. However, the use of these techniques to obtain quantitative structural and energetic information is still limited. We develop experimental, theoretical, and computational methodologies to extend the boundaries of what structural information can be learned using native IM-MS. 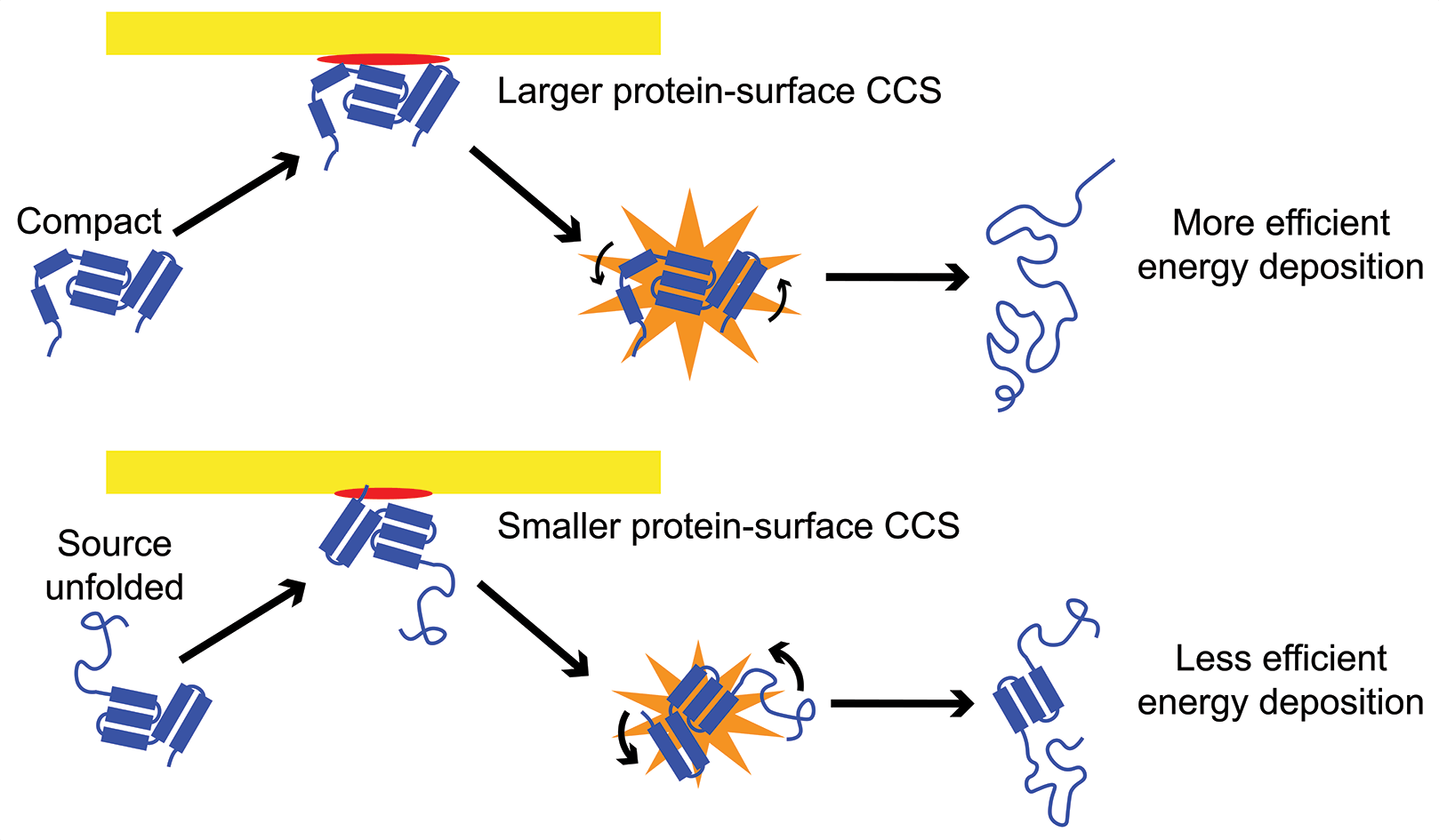 We have constructed a model that simulates energy deposition in CIU in a Waters Synapt G2-Si and used this model to calibrate the energy scale for CIU and show that energy deposition in surface-induced unfolding (SIU) is 20-68% efficient, becoming more efficient for large proteins and high incident kinetic energies. We have also shown that energy deposition in SIU decreases dramatically for more elongated structures, providing insight into the protein-surface interaction. We are also using this model to develop a method to directly determine activation energies for gas-phase unfolding transitions from protein CIU data, enabling facile determination quantitative energetic information. Combining this energetic information with measurements of the change in collisional cross section informs structures constraints for molecular dynamics simulations and provides additional insight into the specific unfolded structures accessed in the gas phase. IonSPA software is also being developed for this purpose.
We have constructed a model that simulates energy deposition in CIU in a Waters Synapt G2-Si and used this model to calibrate the energy scale for CIU and show that energy deposition in surface-induced unfolding (SIU) is 20-68% efficient, becoming more efficient for large proteins and high incident kinetic energies. We have also shown that energy deposition in SIU decreases dramatically for more elongated structures, providing insight into the protein-surface interaction. We are also using this model to develop a method to directly determine activation energies for gas-phase unfolding transitions from protein CIU data, enabling facile determination quantitative energetic information. Combining this energetic information with measurements of the change in collisional cross section informs structures constraints for molecular dynamics simulations and provides additional insight into the specific unfolded structures accessed in the gas phase. IonSPA software is also being developed for this purpose.
Contact persons: Sam Shepherd, Dr. Micah Donor (now at George Fox University)
Funding: NIH R01GM144507, Agilent University Relations Award, (previously) NIH R21AI125804
Membrane Protein-Lipid Interactions and Dynamics
 A major frontier of biochemical and pharmaceutical research is the characterization of membrane protein assemblies, including lipids. Many membrane protein assemblies are believed to associate with dynamic, nanoscale domains of segregated lipids, which may in many cases be essential for the proper function of the assembly. Translocation of proteins and small molecules across lipid membranes is a fundamental process necessary for the function of many toxins, membrane channels, and pharmaceuticals. We develop techniques that exploit the high chemical specificity and sensitivity of native mass spectrometry and ion mobility in order to study the architecture, shape, and lipid interactions of membrane proteins as well as translocation mechanisms.
A major frontier of biochemical and pharmaceutical research is the characterization of membrane protein assemblies, including lipids. Many membrane protein assemblies are believed to associate with dynamic, nanoscale domains of segregated lipids, which may in many cases be essential for the proper function of the assembly. Translocation of proteins and small molecules across lipid membranes is a fundamental process necessary for the function of many toxins, membrane channels, and pharmaceuticals. We develop techniques that exploit the high chemical specificity and sensitivity of native mass spectrometry and ion mobility in order to study the architecture, shape, and lipid interactions of membrane proteins as well as translocation mechanisms.
Many pore-forming bacterial toxins form small transmembrane beta-barrel pores in target cell membranes through which cytotoxic effector proteins can be translocated. Sea anemone alpha-helical pore-forming toxins also comprise a rich diversity of homologs with different pore complex stoichiometries and lipid affinities. We study the shape and structure as well as the lipid preference of these pore assemblies using nanoscale membrane models to transfer them in a native-like conformation into the gas phase. We use a combination of gas-phase fragmentation techniques, collisional cross section measurements, and computational modeling to discover the chemical and physical principles that underlie toxin-lipid assemblies and dynamics.
Contact persons:
Funding: Agilent University Relations Award, (previously) NIH R21AI125804
Fundamentals of Ion Mobility-Mass Spectrometry
 The Prell group develops computational and theoretical tools for understanding and interpreting (native) ion mobility-mass spectrometry data. One of our major projects is called “Collidoscope”, a program that computes likely arrangements of charges on the surface of proteins and protein complexes as well as collisional cross sections in various buffer gases for a variety of ions, from the size of a few atoms up to many megaDaltons. Collidoscope is an open-source, object-oriented program written in C++, and easily parallelizes over many processors and nodes (via the Message Passing Interface system in Linux supercomputing clusters). Collidoscope uses the “Trajectory Method”, which relies on explicit computation of collision gas scattering off of a sum of Lennard-Jones 6-12-4 potentials due to the atoms in the analyte and the collision particle. Collidoscope is available for public use via Github at https://github.com/prellgroup/Collidoscope. We have also investigated the accuracy of protein ion compaction (which occurs as ions are transferred from electrospray droplets into the gas phase due to self-solvation of charge sites by polar groups on the protein’s surface) in native IM-MS.
The Prell group develops computational and theoretical tools for understanding and interpreting (native) ion mobility-mass spectrometry data. One of our major projects is called “Collidoscope”, a program that computes likely arrangements of charges on the surface of proteins and protein complexes as well as collisional cross sections in various buffer gases for a variety of ions, from the size of a few atoms up to many megaDaltons. Collidoscope is an open-source, object-oriented program written in C++, and easily parallelizes over many processors and nodes (via the Message Passing Interface system in Linux supercomputing clusters). Collidoscope uses the “Trajectory Method”, which relies on explicit computation of collision gas scattering off of a sum of Lennard-Jones 6-12-4 potentials due to the atoms in the analyte and the collision particle. Collidoscope is available for public use via Github at https://github.com/prellgroup/Collidoscope. We have also investigated the accuracy of protein ion compaction (which occurs as ions are transferred from electrospray droplets into the gas phase due to self-solvation of charge sites by polar groups on the protein’s surface) in native IM-MS.
Do gas-phase or solution-phase interactions and energetics more strongly influence what is observed in native IM-MS? Another thrust of our group is to use a combination of experiments, theory, and computations to quantitate the strength of gas-phase interactions between proteins and ligands to help better define their potential influence on IM-MS observations.
Contact persons: Jim Prell, Dr. Simon Ewing (now at Intel)
Improvement of Molecular Dynamics Algorithms for Use in Native IM-MS
Simulating large biomolecular ions with MD can be enormously computationally expensive and can often provide misleading results when performed without benchmarking. Which MD simulation protocols provide a good balance between speed and accuracy? To begin addressing this question, our group has investigated 5 different commonly used MD force fields for their accuracy in predicting the typical slight compaction of native biomolecular upon transfer to the gas phase and subsequent desolvation. We found that a simple approach using a readily available GROMOS force field can predict collision cross sections for native protein ions (including charge-reduced and membrane proteins) within 0-4% of experiment. This simulation protocol has become standard in our laboratory and continues to provide excellent agreement with experiments for a large variety of biomolecular ion structures and sizes. Our efforts in this area continue to develop practical, straightforward protocols for predicting and interpreting native IM-MS data.
Contact persons: Jim Prell, Dr. Amber Rolland (now at University of Utrecht)
Funding: NIH R01GM144507, NIH R01EY027012 (PI: Kirsten Lampi, OHSU), NIH R01GM141733 (PIs: Daniel Zuckerman, OHSU, and Elisar Barbar, Oregon State), (previously) NIH R21AI125804